以下はPacBioテクニカルノートに記載している方法です。 (うまく説明できていないかも)
同じゲノム位置の、ベースコールの集合(どれも同じQV)を2つ考えてみます。 1つは100%ワイルドタイプで、もう1つの集合は(50:50または80:20の)変異を持っているとします。 ランダムに2つの集合からベースコールをピックアップするとき、その数はそのゲノム位置でのカバレージと同じ意味です。
2つのピックアップしたサンプルを、Fisherの正確度検定にかけて、どれだけ有意に変異を検出できるか(帰無仮説は、ワイルドタイプの集合からのサンプルも変異がある集合からのサンプルも、差はない)検定をします。
p-valueが0.05未満のとき、有意に検出できるとし、ピックアップする数を1から50に増やしながら(つまりカバレージが1から50のときまで)1000回繰り返します。
このようにしてシュミレーションした結果、(p-valueが0.05未満になるときの数)/1000 = 検出率とすると、0.95以上になるには、
50:50のサンプルでは、CCS-3パス(Q17)のとき17X、CLRフルパス(Q8.24)のとき29X必要で、
80:20のサンプルでは、CCS3-パスのとき55X、CLRフルパスのとき135X以上必要であることがわかりました。
この数値はもちろん理論値です!
さて、以上をふまえて、ターゲットサイズ(アンプリコンのサイズ、インサートのサイズ)ごとに、SNP Detectionに必要なSMRT Cellの数を計算した表があります。
計算式はこちら

で、tはターゲットの数、cは最低必要なカバレージ(先の理論値より若干高めに設定)、βはサンプルバイアス(N倍くらい多めに読まないと期待される変異は取れないんじゃないか、という値)で3に設定。 tcβで必要リード数が計算されますので、それを前回の書き込みで紹介した、Cellごとの、ある程度クオリティが期待される「使える」リード数=rで割ると、必要Cell数が計算されます。


(Table4の20% variantの500bpの、Estimated # SMRT Cellは2ではなくて3が正しい)
この2つの表からわかることは、500bpのターゲットでは、ターゲットの数が500のときCLRとCCSはどちらも1つくらいしかCellを必要としない、ということ。
でも、この表には無いですが、ターゲットの数を500から1000、1500、2000と増やしていったらどうなるのか、上記の式に代入して計算してみました。
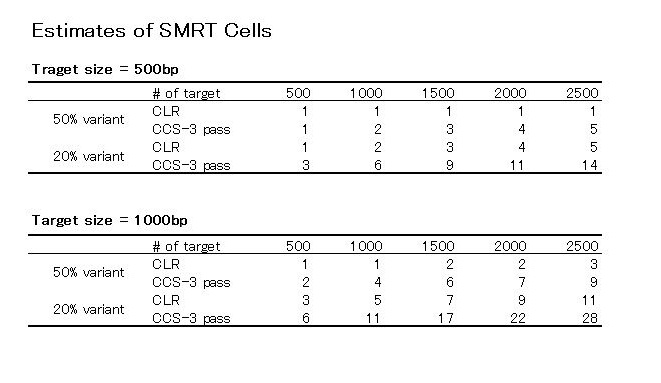
ターゲットサイズ500bpのときと1000bpのときの2パターンでCLRとCCSのどちらがコスパがいいか?
ターゲット数が増えるほど、CLRでやったほうが、変異割合が50%のときも20%のときも少ないCell数で済む、と言えそうです。
Again, あくまで理論値です!
では実験値ではどうなのか。
Agilent SureSelectを使ったターゲットエンリッチメントの例があります。
続く



