ブログを書く暇があるので、今日は、最近ウェブで紹介された、メディカル関係の記事を3つ紹介します。
まずはこちら、Diagnostics Worldからの記事
Long-Read Sequencing in the Age of Genomic Medicine

Icahn School of Medicine at Mount SinaiのBobby Sebra氏曰く、「Pseudogenes, large structural variants, validation, repeat disorders, polymorphic regions of the genome―all those are categories where you practically need PacBio」
ヒトゲノムに存在する変異には、ショートリードでは検出できない大きな構造変異、リピート変異が多く存在します。これらを検出するにはPacBioのロングリードが不可欠、という話。
技術はある。マシーンもある。あとはソフトだけ。
ソフトウェアは、研究者向けには一通り揃っていますが、お世辞にも現場の医者向けではありません。
それはどのNGS機器でも同じ問題を抱えているでしょう。
Mount Sinaiでも、3台のPacBio RSIIから出てくるデータを効率よく解析できる様に、彼ら独自のソフトウェア・アルゴリズムを作っています。(例:構造変異検出ツールとか)
また、ひとつのテクノロジーにこだわらず、10Xや、BioNano、Oxford Nanoporeなどいろいろな技術を次々に取り入れ試しています。
最近2倍体ヒトゲノムをPacBioとBioNanoで解析し、それまでのどの結果より優れたContig/Scaffold配列を作りました。
このように、他のNGSとは明らかに優位性を持つPacBioのロングリード。
メディカルの現場に持ってくるにはどうしたら良いか?
言うまでも無く、機械の使いやすさと安定性、低コスト、わかりやすいソフトウェア、が必要でしょう。
RSIIからSequelになって、実際のところはどうでしょうか?
Sebra氏は、Sequelのアーリーアクセスユーザでもあります。
その辺の話は、また今度、2月23日(火)に秋葉原で行なわれる、「第二回PacBio現場の会」ワークショップセミナーにて、詳しく聞けますよ。
お時間があれば是非ご参加下さい!お楽しみに!
ちなみにこの記事の中に登場する英語で、「Off-the-shelf」というのがあります。
いつでも買える、とか、入手可能な、という意味です。
Off-the-shelf analysis, with readable diagnostic reports for doctors
というと、医者が読みやすい、臨床レポートが出てくる解析ツールの存在、という意味。
今はまだありませんが、これなくしてクリニカルシークエンスの現場に普及するのは難しいですから、絶対できるはずです。
次はGenomeWebから(ごめんなさい、これは有料みたいです)
Baylor Team Explores PacBio Long Reads for Detecting Pathogenic Structural Variants in Patients
こちらはBaylor College of Medicine & Miraka が考えている、ヒト診断への可能性についての記事です。
BaylorのAssistant Prof. Bainbrigdeは述べています(ざっくり翻訳)。
「ヒト全ゲノムのシークエンスが現実的なものになり、皆さんこれで構造変異などの問題は全て解決できる!と思ったはずです。でも、たいていの構造変異検出プログラムは、変異全体の1/3~1/4ほどしか検出できていないんです。それらを合わせても半分くらいしか実は検出できていないことになります。でも、6,000~10,000 bpのロングリードを使えば、もっと簡単にできるはず」
Proof-of-Concept(できるかどうか試してみよう実験)にて、Baylorのチームは、PacBioのロングリードが、どれだけ遺伝子検査に応用できるかを調べました。
彼らはまず、既知の疾患関連遺伝子ベスト100にフォーカスしています。その中には、がん関連遺伝子60余りや、シングルセキソンの抜け落ちが原因で引き起こされる疾患関連遺伝子、Fragile Xシンドロームなどリピートの数が重要となる遺伝子、が含まれます。
そのような、重要遺伝子だけをキャプチャーする技術、キャプチャーシークエンスとかターゲットリシークエンスとか呼ばれますが、これをPacBioのロングリードにも応用できます。
前のブログ「PacBio-LITS PacBioでターゲットリシークエンス 1& 2」でも紹介しましたが、プローブを用いて、7kb程度のロングリードをキャプチャーできるようになったのです。
今までのキャプチャーシークエンスといえば、タンパクコーディング箇所に焦点を当てた、エキソームシークエンスが有名ですね。
各メーカーがキットを出しています。
PacBioのロングリードでできるキャプチャーは、エキソンのみならず、遺伝子全体なんです。
遺伝子全体をキャプチャーできるアプリケーションはPacBioのロングリード無しではできない。
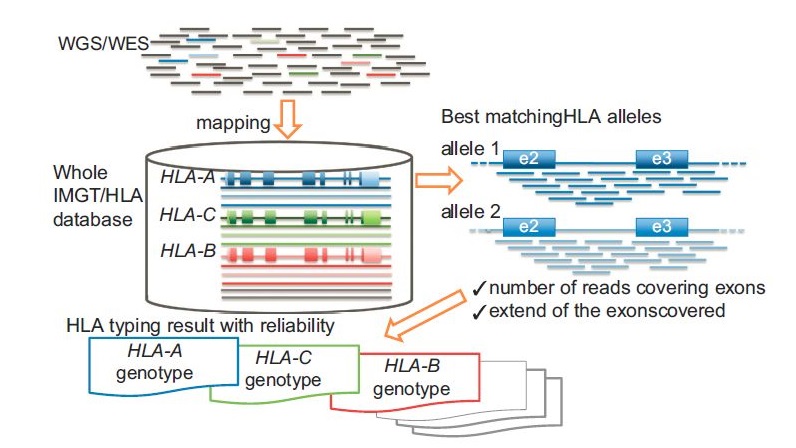
これはがん遺伝子もそうですが、HLA遺伝子なんかも、全体をキャプチャーしたほうが複雑な変異(phasingなど)を検出できるので良いと思います。
最後は、こちら
Front Line Genomics Magazine Issue Six

こちらの雑誌はフリーで全文読むことができます。
その42~43ページめにPacBioの記事があります。
インタビューに答えているのは、PacBio本社のマーケ担当、イケメンLuke Hickey
内容は、これまでPacBioを使って挑戦されてきたヒトゲノムアセンブリの紹介と、On-Goingなプロジェクト。
ハプロイドのヒトゲノム(hydatidiform mole, CHM1)をPacBioで読んで読んで・・・というのは、以前このブログでも紹介しましたが、その時はWashington University in St. Louisのプロジェクト。
大学名が似ているのでたまにごっちゃになるのですが、こちらはthe University of Washington、さらにUniversity of Bari Aldo Moro と University of Pittsburghのチームは、同じくハプロイド株を40X PacBioで読んで、リファレンスゲノムに存在する160箇所のギャップのうち半数以上を、クローズするかギャップを縮めたそうです。
そのほとんどはGCリッチな場所、反復領域だったとのこと。
さらに、100万塩基以上の新規の配列を追加。
彼らが見つけた26,079個の変異箇所のうち、コピー数変異の85%、挿入変異の92%、欠損変異の69%が、これまでに報告の無い新規だったそうです。
ということは、リファレンス配列といっても、まだまだ完璧ではなかったんですねえ。
リファレンス配列といえば、ここ数年、人種ごとにリファレンスを作ろう!的なプロジェクトが動いています。
韓国のマクロジェン社とソウル大学が推し進める、韓国人リファレンスゲノム計画は、PacBio、BioNano、BACシークエンスを使ってデータを出し、完成に近づいています。
GRC(Genome Reference Consortium)アセンブリよりも、よりアジア人を代表するリファレンスゲノムになる予定です。
さらに、あのCraig Venter博士が率いるHuman Longevity, Inc. では、世界の人種を代表するゲノム、30リファレンスを作成する計画があるそうです!
何とも壮大な計画・・・
 |
| Front Line Genomics Magazine, Issue Six, p43 |
こういう夢のある計画、いいですね。
ヒト以外の生物でも、もう一回PacBioで全ゲノム読んでみたら、意外と新規配列がたくさん見つかるかも。