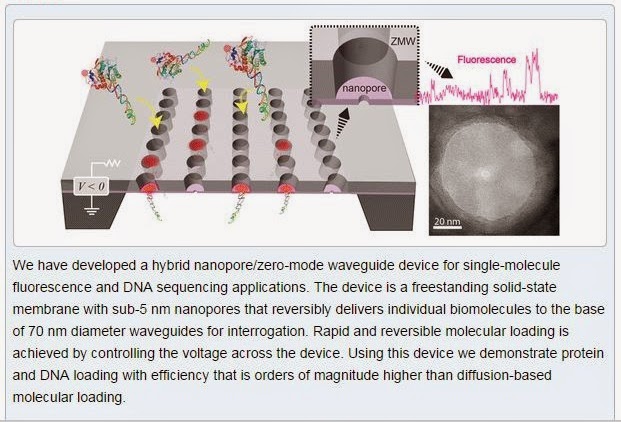
何度も言いますが、初心者向けに作られた内容ですので、物足りないひともいるでしょう。
まあ、お許し下さいませ

タイトル:PacBioRS II によるアイソフォームシークエンス

<概要>
Pacific Biosciences社のDNA一分子リアルタイムシークエンサー、PacBio RSIIは、これまでのシークエンサーの常識を超える、ロングリードを出力する。これまで、次世代シークエンサーで発現遺伝子のアイソフォームを把握するには、出力されるリードが数百bpと短いため、読んだ後にアセンブルをする必要があった。しかしこの方法では、リードがある遺伝子のどのアイソフォームを読んだものかを正しく知ることは難しい。一方PacBio RSIIは、数千bpにわたるロングリードをハイスループットで出力できるため、転写産物のcDNAを、5'側から3'側まで一気に読むことができる。これにより、アセンブリ無しで、完全長のアイソフォーム配列を得ることが可能になった。
<本文>
完全長cDNAアイソフォームシークエンス
PacBioのシークエンステクノロジーは、最新試薬P6-C4で平均10Kbpのリードを出力することができる。このロングリードの威力を発揮するアプリケーションのひとつは、ロングアンプリコンシークエンスである。Alternative Splicingによって様々なアイソフォームを持つ転写産物を、完全長cDNAにした後にPacBioシークエンス用にライブラリを作製して読めば、完全な形でアイソフォームの配列を読み取ることが可能である。
既にアメリカ・スタンフォード大学やワシントン大学などでは、ヒトのトランスクリプトームを完全長cDNAで読むことで、新規な転写産物をたくさん報告している(Sharon et al. (2013) Nat Biotechnol doi: 10.1038/nbt.2705、Au et al. (2013) PNAS doi: 10.1038/pnas.1320101110.)。
 |
| 図1.アイソフォームの完全配列から、PacBioのライブラリを作製してシークエンス |
ライブラリ作製の流れ
PacBio RSIIを使ったアイソフォームシークエンスで良いデータを出すためには、完全長cDNAの作製、ライブラリのサイズセレクション、という2つの重要なステップがある。これらのステップを正確に行うことが、成功への鍵である。
最初のステップである完全長cDNAの作製には、タカラ・クロンテック社のSMARTerをお勧めしている。これは、mRNAのPoly‐A側にオリゴdTプライマーにより逆転写を行う。比較的簡便に、完全長のcDNAを得ることができる。ほかにも完全長cDNAを作製するキットは他社から販売されていて、PacBio本社のラボでもテストが行われている。
完全長cDNAが作製されたら、PCR増幅し、アンプリコンを作る。このときのPCRサイクル数は、必要最低限に抑えるのがコツである。過剰に増幅されるバンドが無く、どのサイズも一定に増えるようにサイクルを最適化する。
次に増幅したアイソフォームのアンプリコンは、サイズを分けて回収する。サイズは例えば1-2kbp、2-3kbp、3-6kbpというふうに3つに分ける。サイズを分けて回収したそれぞれのサンプルで、別々にライブラリを作製する。サイズを分ける理由は、短いライブラリと長いライブラリを混ぜてシークエンスした場合、短いライブラリが長いライブラリに比べて優先的にシークエンスされやすい、というPacBio RSIIシークエンサーのウェル独自の特徴があるためである。サイズセレクションは、ゲルで流してマニュアルで分画するか、Sage Science社のBlue PippinやELFのような自動分画装置を使用する。
ここから先は、PacBio社のキットを用いて、DNAの損傷修復、平滑末端化を行う。そして、両端にヘアピンアダプターをライゲーションする。こうしてできたSMRTbellライブラリは、プライマーをアニーリングさせ、ポリメラーゼを結合させてからPacBio RS IIシークエンサーにセットし、サイズの異なるライブラリごとに、別々のセルでシークエンスする。
バイオインフォマティクス
PacBio RSIIから出力される生リードは、最新試薬P6-C4を使った場合、平均10kbpに達する。一方、cDNAの完全長はたいてい1.5kbから長くても5kbpが普通である。一見するとPacBioのリードは長すぎるように見える。しかし完全長のcDNAを高精度に読むためには、できるだけ長いリードが出力される方が望ましい。理由を以下に示す。
図2に示すとおり、PacBioのSMRTBellライブラリは、そのダンベル型の構造によって同じ配列を何度も読むことができる。インサートが短いcDNAの場合、この箇所を何回も読むことにより、1回で読んだ場合のエラーを自ら補正することができる。PacBioの生リードは精度が86%程度であるが、エラーの多くがランダムなInDelなので、同じ分子を何度も読んだときに得られる配列を重ね合わせることで、エラーをキャンセルし、精度を99%以上に高めることが可能である。
このようにして得られた1分子DNA由来のコンセンサス配列のことを、Circular Consensus Sequence (CCS)、またはReads of Insert (ROI)と呼んでいる。
コンセンサス配列は次に、フルパスのcDNAを含むか否か、に分けられる。フルパスのcDNAは、5'側プライマーとUTR、コーディング領域、Poly-A配列、3'側プライマーを含む。確実にフルパスが読めていたものだけを抽出する。
プライマー配列とPoly-A配列をトリミングした後、同じアイソフォーム由来のコンセンサス配列をクラスタリング手法を使ってマージする。このようにしてデータの冗長性を取り除きながら、同時に完全長cDNA配列の精度を上げる。
最後に、生データが持つパルスデータなどの情報を使用して、再度配列の精度を上げる。このようなプロセスを経て、精度99%をはるかに越える、正確な完全長アイソフォーム配列が完成する。
リファレンスゲノム、遺伝子アノテーションがある場合は、アイソフォーム配列をゲノム配列にマッピングすることで新規のアイソフォーム配列を見つけることができる。
これら一連の流れ、フルパスのコンセンサス配列の抽出、クラスタリングから精度向上アルゴリズムまでは、PacBioの解析ソフトウェア-SMRT Analysisで簡便に行うことができる。
アイソフォームシークエンスの特徴
PacBioの超ロングリードによる完全長cDNAシークエンスの目的は、一分子由来のアイソフォームを完全に読みきることで、Alternative Splicingの異なるアイソフォーム配列を検出することである。最終的な出力配列からは、量的な情報(発現量の情報)は除かれている。アイソフォームごとの発現量を解析するには、ショートリードシークエンサーのデータを組み合わせるなど、工夫が必要である。
 |
| 図2、1分子のcDNA全長は、PacBioのロングリードで何回も読まれる |