11日のランチョンは、PacBioのプレゼンということで、満席でした。 良かった良かった。
講演を聞いた方はご存じでしょうが、PacBioのロングリードは平均15%のエラーがあります。 そこでショートリードでマッピングして、エラーをできるだけ取り除き、精度を増したロングリードでアセンブルすると、それ以外のどの組み合わせよりもN50、最長Contigなどで成績が良かったとのことです。
このグラフ↓は、その時のプレゼンでも紹介されました。

PBcRは、PacBio correction Read で、エラーコレクションがされたロングリードです。
左から3つは、PacのCCSのみ50X、IlluminaショートリードPaired Endの100X、454の50Xです。
リード長はどれも数百塩基です。
これらはどれもN50が100Kb、Max 300Kb程度ですね。
次いでCCSでエラーコレクションしたロングリード(どちらも25X)、454でエラーコレクションしたロングリード(どちらも25X)。 この2つはほぼ同じような成績で、N50が200-300Kb、Max 600Kbでした。
Illuminaでエラーコレクションしたロングリード(どちらも50X)はN50が300Kb、Max 1Mbで、一番良かったのがCCSでエラーコレクションしたロングリードの50Xで、N50が500Kb、Max 1.2Mb
細かい数値やメソッドは、近く論文として紹介できると思います。
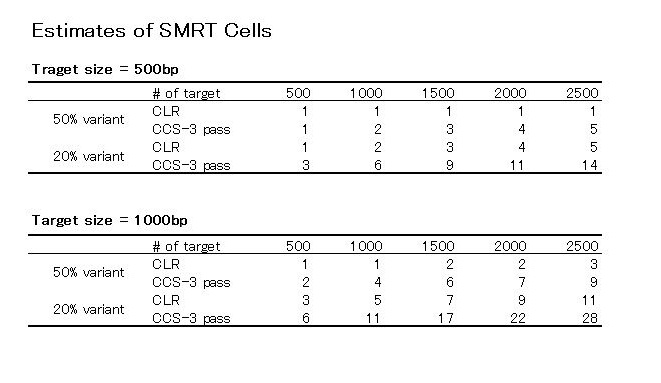
Pacのデータを使ってできるアセンブリの種類ですが、大きく3つ、あります。
- SMRT Hybrid
- SMRT Scaffolding
- SMRT de novo
SMRT Scaffoldingは、精度が高くなったContigやロングリードを使って、Contig間をつなぐ(Scaffoldingする)こと。
SMRT de novo は、Pacのロングリードだけをそのまま使ってアセンブルすること。
です。
SMRT Hybridは一番クオリティが高いアセンブル手法です。 Illuminaや454データを持っている場合やCCSを組み合わせて実験したデータを持っている必要があります。
ゲノムサイズに制限の無いCelera Assemblerがお勧め、とのことです。
ALLPATHS-LGというアセンブラーもありますが、こちらは10Mbまでという制限があります。
制限といえば、Celera Assemblerも、インプットするロングリードの個々のリード長が30Kbを超えてはダメという制限がありますが、今のところPacのロングリードも30Kbは読めませんので問題無いでしょう。
将来的には・・・ 30Kb読めてしまうかも
SMRT Scaffoldingは、ショートリードで作ったContigがある場合や、PacのCCSなどで作ったContig、Pacのロングリードをエラーコレクションしたリードがある場合、それらのContigの精度が高いと見なされる場合に、Contig同士をブリッジするのに有効です。
(Scaffoldingのちょうど良い日本語訳が見つからない!)
SMRT de novo は、ゲノムサイズが1Mb未満の小さいゲノムを、CCSを使わずに、ロングリードだけで読んで、クオリティは若干低くてもいいからドラフトゲノムを作りたいときなどに向いています。
ウイルスなどは少ないCellでもカバレージも十分とれるので良いかもしれません。
以上のアルゴリズムについては、後々、書いていくつもりです。 っていうか、
書かざるを得ないでしょうねえ。