ナノポア、といえば真っ先にOxford Nanoporeが頭に浮かぶひとも多いでしょうが、ナノポアとはナノテクノロジーで作る穴、の一般名称
Oxford Nanopore以外にも、日本ではクオンタムバイオシステムズとか日立ハイテクノロジーズとかが独自、あるいは海外の会社と共同で開発している。
し か し、
今日紹介したいのは、米国・ボストンにあるNortheastern University
ここのLarkin博士らは、PacBioと共同で、SMRT CellのZMWに小さな穴「ナノポア」を開けて、微弱な電圧をコントロールすることで、DNAを可逆的にローディングし、スループットを飛躍的に向上させる可能性があることにR&Dレベルで成功した。
回りくどい言い方だが、そう、彼らはローディングをUpさせることに成功したのであって、シークエンススループットがUpしたとは言っていない。まだ。
次のチャレンジだ、と言っている。
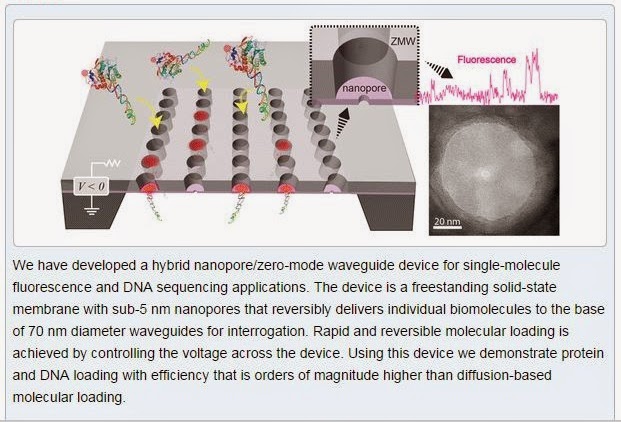
J Larkin et al., (2014) Nano Letters DOI: 10.1021/nl503134x
彼らは、ZMWの底、直径70nmの面に(これがガラスではなく窒化ケイ素の膜になっている)、TEMで2.5~4 nmの穴を開けた。
そして、ビオチンのついたDNAと、蛍光ラベルされたストレプトアビジンたんぱく質、これをひとつのNZMWに投入、電圧を操作した。
高電圧下では、ストレプトアビジンはDNAから離れ、電圧を逆にすると、ストレプトアビジンたんぱく質-DNAの複合体はNZMWから放たれた。
さらに彼らは、8つのNZMWに、DNA-ストレプトアビジン複合体を流して、電圧を操作すると、8個中5個のNZMWから蛍光を検出した。
つまり62%のローディング効率、(我々の専門用語では「P1が62%」という!)
8個中5個で62%ねえ・・・。
今のマグビーズローディングでは、大体、20%~良くて50%といったところ。
平均は30~40%かなあ。
62%は確かにローディングの数字だけを見ればすごい。
しかしまだ、これはDNAを読んだわけでは無いし、15万の全ZMWを対象にした話でも無い。
技術としては面白いと思いますよ。
物理の得意なひとは、論文読んでみてください。
実用化したら、DNAは今の半分の量で済むかもしれない
DNA分子の動きをコントロールできれば、もっと効率的にシークエンスできるかもしれない
ああ、そういえば、DNA分子の動きをコントロールすることが、ナノポアシークエンス技術の最も難しいところだと、昨年京都の一分子DNAシークエンサーシンポジウムで何人も言っていたなあ。
DNAは3次元の構造体だから。
しかし、世界には面白いことを思いつくひとがいるんですね。
半導体やナノテクの技術者からは、生物やっているひとには想像も付かないアイデアが出てくるのかも。
interdisciplinaryという言葉を知っていますか?
日本語では、「多分野にまたがる」、「諸学連携」、という意味です。
色んな分野の専門家が集まったほうが、イノベーションを生むんですよね。
企業もそうであるべきだ、と最近思います。
最後にこの、Nanopore ZMW、略してNZMWは、もちろんまだ製品化されていないし、予定も無い、そうです!













