その時例に使ったデータは何かと言うと、Sequelでヒトゲノム NA12878 を10xくらいの深度で読んだものです。
具体的には、ここのデータ

- 使用したSMRT Cell 1M の数:10
- トータルラン時間:60時間
- 出力塩基数:32.8 Gb
- リード数:340万本
- リード長のN50 :11.823 bp
このとき使用した試薬は、旧バージョン、v.1.2 のもの
なので今ならセルあたりの出力はもっと多いはず。
とにかくこれでヒトゲノムの10倍のデータが出た。
このデータをヒトゲノムにマップするのですが、ここで使ったツールは、NGM-LR + PBHoney
PBHoneyは構造変異を検出するツールです。
NGM-LR って何? という方、これはロングリード用のマッピングツールです。
Next-Gen Mapping tool for Long Read、だったかな?何かそんな名前。
Githubにもあるので、興味のある方はここからどうぞ。
PacBioリードは1本が長いので、例えば 1kb 程度の挿入・欠損をまたいで読むことが可能。
しかし通常のマッパーでは 1kb の変異を考慮してほかの配列を綺麗にゲノムにマップすることができなかった。NGM-LRは、二箇所に分かれてマップするような、ロングリード独特な性質をフルに発揮できるマッピングツール。
 |
| BWAとNGM-LRのマッピング結果 Aaronのスライドより |
先ず、DNAnexusのデモアカウントと作りましょう!
いえいえ、決して私はDNAnexusの手先ではありません。
仕方無いんです。ここにアクセスした方が、データ参照が楽だから。

私はアカウント持っているのですが、ロングインするとこんな感じです。

左下の、"PacBio Sequel Data" というところをクリックします。
これが例の10カバレッジのSequelデータ


そこで、indexsession ファイルをダウンロードします。
これは、IGVに取り込むと、DNAnexusのサーバにアクセスしてデータを表示してくれるインデックスファイルです。
IGVはこのように、必ずしもローカルに大きなサイズのマッピングファイルを持っておく必要がありません。
さ、IGV を開きましょう。前回のあれ、ですよ。わからないひとはちょうどこの前の記事をチェック!


File > Open Sessions
今ダウンロードした indexsession ファイルを選択。
何か聞かれるけどOKをする。

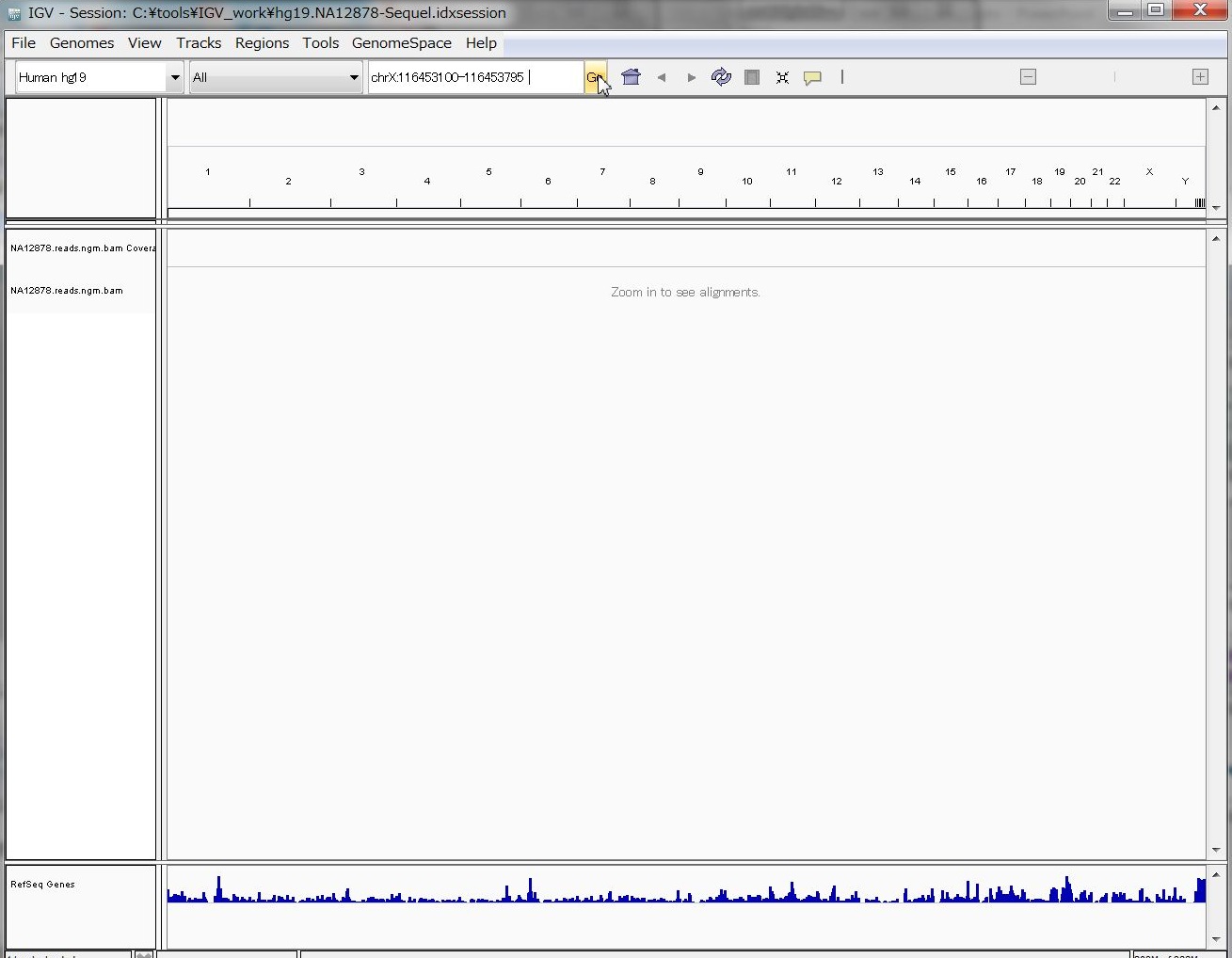
ゲノムのポジションを入力する場所に、試しに、
chrX:116453100-116453795
と入れてGo!

いぇーーーーい !!
ほかにもチラッと見てみたいポジションは、こんなところかな?
chrX:116454160-116454859chr10:92213800-92216245
InDelの変異箇所は、先の DNAnexusのアカウントから、
NA12978_Output を選び、ここから ... del.bed ins.bed ファイルをダウンロードしてきて、取り込むと面白いかも!

いかがだったでしょうか?




















