私が愛読しているブログに面白い記事がありました。
「PacBioでMicrobeをシークエンスするコスパは?」
(原文:The economics of PacBio sequencing a microbe)
JGI(Joint Genome Institute)ではPacBioとHiSeqを保有しています。
そこでは、バクテリアや少し大きめ真核生物ゲノムをシークエンスしています。
早くからPacBioRSを導入しただけあって、いろんなプロトコル開発などにも協力してきたみたいです。
さて、バクテリアや真核生物ゲノムのドラフトを決めるのに、全部PacBioで読んでしまえば、それは精度の高いものができるはずですが、コストがHiSeqと比べてまだ10倍するそうです。
ですので、ラフでも良いものや、単純なゲノム構造の生物は、2x150bp, 275bpインサートライブラリを、Illumina 1T HiSeq 2500で、バクテリアは48x のマルチプレックス、真菌クラスは8xのマルチプレックスで読んでいると。
もちろん、GCが極端に多かったり少なかったりするゲノムの場合、ショートリードだけではつながらないので、そこでPacBioの登場!
例えばGCが70%を超えて、数キロにわたる長ーいリピート配列を含む放線菌の場合、ライブラリを作ってAMPureビーズによる10kb以上のセレクションを行い、PacBioRSIIで読めば、たいてい2-3個のSMRT Cellでゲノムアセンブリは完了するとのこと。
この辺は我々の認識とほぼ合ってます。
JGIは昨年度、250株ほどを読んだらしいです。今年は500株にのぼるとのこと。
真核小型ゲノムの場合、5個のSMRT Cellでおおよそ40Mbのハプロイドであれば、かなりちゃんと読める模様。
10kbのAMPure精製カットオフをしたシークエンスということですから、妥当な数でしょう。
今期はBluePippinを購入、20kbライブラリプロトコルでもっと複雑なゲノムにも挑戦です。
さて、真核で小型ゲノムでは、4kbのメイトペアライブラリを作製し、ALLPATH-LGでアセンブルする方法もとっているそうですが、バクテリアではそれをしていない。
なぜかと言うと、メイトペアライブラリを作るコストと、PacBio RSIIで1セル読むコストがほぼ同じで、かつPacBioの方がアセンブリ結果が良いから、だそうです。
同じ理由で、今後は真核生物でもPacBioだけで読むことになるらしい。
さて、コストと言えば、新型機種・Sequelシステムは、ランニングコストはRSIIの2~3倍、と先のブログには書いてあります。
日本定価ベースで比較すると、SequelのランニングコストはRSIIの1.6倍~3.6倍です、
これにはサンプル調整からシークエンス消耗品まで全て含んだ、より実感に近いのランニングコストでしょう。
ま、2~3倍というのは間違いではないですね。
Sequelの場合はセルが高価なので、セルが増えれば、コストは上がります。
但し、Sequelのスループットは公表値でRSIIの7倍
ギガベースあたりのコストはSequelの方が4倍~8倍安くなる計算
安くなるとは言っても、スループットが多いというのもまた、微生物やっている研究者には考えもの。
というのも、バクテリアでゲノムサイズが5Mb 程度で、今までRSIIでセル1個で読めていたものを、わざわざSequelで読むかという問題があります。
ここも日本定価で換算すると、RSIIで3個のセルを読む場合と、Sequelで1個のセルを読む場合と、ほぼ同じ(まだ若干Sequelの方が高い)です。
なので、RSIIでセル4個以上でないと読めないくらい複雑なゲノムであれば、Sequelで読んだほうがお得感が感じられるかもしれませんね。
それでもまだオーバースペックであれば、バーコード(マルチプレックス)という方法があります。
先のブログでは、例えばRSIIで1セルで十分読めるようなバクテリアのライブラリの場合、7種類のバーコード付きライブラリをSrequelのセル1つで読めば(7倍のスループットなのだから)、コストを大幅に抑えられる、というような文章があります。
ちょっと誤解があるのですが、バーコードを付けると、リードの出力は減ります。
バーコードは、2本鎖DNA(インサートDNA)の末端とヘアピンアダプターとの間にあります。
ですので、シークエンスが始まってから、アダプターまで届かなかったリードはそもそもバーコード配列が読まれません。それらはポリメラーゼリード、サブリードとしては出力されますが、バーコード配列を基準に分けることができない。
これはライブラリが長くなればなるほど、バーコード配列まで読まれないリードが多くなる、ということを意味します。
今は、デノボアセンブリ用のアダプターバーコードは、10kbライブラリ用で使うことを推奨しています。 しかーし、それでもバーコード認識できないリードが、多くて全体の50%ほどあるかと思います。(平均リード長=10kb なので)
そんなわけで、どうしてもオーバースペックに感じられる場合は、バーコード無しで複数ライブラリを混ぜてシークエンスし、後で一緒にアセンブルする、という少々乱暴なやり方も存在します。
この方法は、サンプル間のゲノム配列が全く異なる場合にのみ有効です。
相同性が高い株同士を一緒にアセンブルすることは、ミスアセンブルを導くだけなのでやめたほうが無難。
JGIではそのようなことが議論されていたようです。
私も今年の春に、海外のユーザで同じように、完全に異なるBACをバーコード付けずにシークエンスして、完全に元通りにアセンブルが再現できた例を聞きましたし、
PacBioのUS本社でも半ば常識のように話されていた記憶があります。
いずれにしても、Sequelは、出力データ量(正確には塩基数)が7倍ということですので、小さいゲノムをアセンブルするときには工夫する必要がありそうです。
データ量が様々なサイズのセルが出てくれれば良いですね。
ついでに記しておくと、先の引用ブログの、装置の値段は正しくありません。
300kドルというのは間違い。そんなに安かったら嬉しいですけど。
2015年11月6日金曜日
2015年10月10日土曜日
ASHG2015
-------以下の記事に誤字・脱字がありましたので訂正するとともに、最後のほう、少し文章も変えました------
今年もアメリカ人類遺伝学会に行ってきました。
場所はボルチモア。ここは、歴史があるきれいな街並みでした。
夜は気をつけないといけない、のはアメリカの都市ならでは、かな。
さて、今年もいろいろ話題はあったと思いますが、私が気になったのは、いよいよNGSが臨床に現場に使われてきたということ。
2、3年前から何となく、クリニカルシークエンスとか言われていましたが、やってみて、いろんな問題点が見つかった。
そのひとつは、ゲノムのリファレンス配列が無いということ。
人種ごとのリファレンスが無い。
これは今までも言われてきたことですが、研究ではなく、クリニカルに用いるためには、高精度のリファレンス配列が必要です。
そのための技術、が、3年前は無かったに等しい。
そして今、ようやく技術が出揃ってきて、ヒトのハプロイドゲノム(いわゆるプラチナゲノムCHM1とかCHM13とか)や、Diploidゲノムのリファレンス作りが可能になってきました。
今回、高精度のリファレンスを作ろう! というときに必ずといって良いほど出てきたテクノロジーが、PacBioとBioNano
これは前にも書きましたが、



今年もアメリカ人類遺伝学会に行ってきました。
場所はボルチモア。ここは、歴史があるきれいな街並みでした。
夜は気をつけないといけない、のはアメリカの都市ならでは、かな。
さて、今年もいろいろ話題はあったと思いますが、私が気になったのは、いよいよNGSが臨床に現場に使われてきたということ。
2、3年前から何となく、クリニカルシークエンスとか言われていましたが、やってみて、いろんな問題点が見つかった。
そのひとつは、ゲノムのリファレンス配列が無いということ。
人種ごとのリファレンスが無い。
これは今までも言われてきたことですが、研究ではなく、クリニカルに用いるためには、高精度のリファレンス配列が必要です。
そのための技術、が、3年前は無かったに等しい。
そして今、ようやく技術が出揃ってきて、ヒトのハプロイドゲノム(いわゆるプラチナゲノムCHM1とかCHM13とか)や、Diploidゲノムのリファレンス作りが可能になってきました。
今回、高精度のリファレンスを作ろう! というときに必ずといって良いほど出てきたテクノロジーが、PacBioとBioNano
これは前にも書きましたが、
- PacBioで読んでアセンブルして、
- できたContigとBioNanoのMapデータをHybrid Assemblyして、
- すごく長い、Scaffoldを作り、
- さらにPacBioでGapを埋めて
- プラチナレベルのリファレンスを作る
というものです。
これをやろうとすると、どれだけ読めば良いか?
いくつかセッションを聞いた話や、実際PacBioやBioNanoの方とディスカッションして聞いたところ、
PacBioは30x~60x
BioNanoは60~90x
これくらい読めば、高精度の長いアセンブリ(一例としてはCHM13の場合、Contig数254、N50=20.79Mb、最長Contig=83Mb)が実現できるとのことです

まさに、シークエンスとPhysical Mappingの融合
PacBioもシークエンスレベルでは素晴らしい成果を出します。
でも、100kb以上の大きなSegmental DuplicationやInversionはさすがに読みぬくことは困難。
そこで、Physical MappingのBioNanoの登場、というわけです!
上記のスライドは、BioNanoのディナーパーティにて発表されたプレゼン。
Physical Mappingがゲノム決定にいかに大事か、ということがわかりました。
間もなく、世界のいろいろなところから、人種ごとのリファレンス、それもかなり高精度のリファレンスゲノムが発表されるでしょう。プラチナレベルで。
やっぱり大事なんですね、ゲノムは。
日本でも大型ゲノムを、PacBioとBioNanoを駆使して、ハイブリッドアセンブリした発表が出ないかなあ、と思う今日この頃
さて、PacBioのブースは最近4年間のASHGで一番大きなものでした。

初日は、PacBio創始者の1人、Steve Turner氏、PacBioのCEO Mike Hunkapiller氏、も勢ぞろい。ブースは常に人大杉状態!
そんな中この、新型マシンの問い合わせはひっきりなしでした。

あ、そうそう、BioNanoのIrysも、スループットが2倍になったそうです。
これで、PacBioのデータとBioNanoのデータを使ったHybrid Assembly の解析を誰でもできるようになったということ。
じゃあ、実際解析はどこでどうするの?
そんなときは、DNANexus !!
このクラウドソルーション型のNGS解析サービスは、既に、PacBioとBioNanoのHybrid Assemblyパイプラインを装備。
クリック&ドラッグ、ボタンをぽち
これでヒトのアセンブリ→BioNanoとのアセンブリ、バリアントコースまでが、2週間で終了!!!だそうです。
すごくないですか? 計算資源で困っている方はこういうクラウドサービスを利用するもの手ですね。
私もDNAnexusの方に知り合いは多いので、何かあれば連絡下さい。
他にも解析サービス会社はあると思うので今度シェアします。
あーあ、来週は日本での人類遺伝学会か。
Sequelの日本での反応はどうかな?
私は2日目から本格参加します。
15日はBioJapanのほうに出ます。
こちらでも、PacBio関連の発表がありますよ。
BioJapanに行かれる方は、是非チェックしてみて下さい。
2015年10月5日月曜日
40kbライブラリ&6時間シークエンス最強説
「40kbライブラリと6時間シークエンス」で、驚きのアセンブル結果!
本当は10月最初の話題はこれにするつもりだったのですが、新型機械の発表があったもんだから、インパクトに欠けてしまいました。
でも、8月に実際シークエンスをしてみて、アセンブルしてびっくりしたのでせっかくだからシェアします。
(公開にあたりサンプル提供者の許可は得ています)
このサンプル、腸管出血性大腸菌O111は、ゲノムの中に20kb~30kbのリピートが多く、ショートリードではアセンブルがとても難しい。
PacBioでも、一昔前の酵素では、同じように複雑なゲノムであるO157:H7ゲノムは200カバレッジで読んでもContig数は9本でした。
(Koren S., et. al. (2013) Reducing assembly complexity of microbial genomes with single molecule sequencing. Genome Biology, 14:R101 Table 3)
そんなところに、この度、最長Movie時間が4時間から6時間にバージョンアップ!
6時間シークエンスということは、超長いライブラリを作成すれば、長いサブリードが得られ、結果アセンブル結果も改善できるはず。
ということで、
を試しました。
40kbライブラリを作製するには、それなりに長くゲノムを切ることが必要。
今までのG-tubeではなく、Megaruptorという機器を使って切りました。
Megaruptorについてはこちら
この機械で何回かテストカットして、Pippin Pulseに流して確認。
本番カットでゲノムを40kb Shearingしたら、PacBio SMRT bellライブラリ作製へ。
ライブラリができたらおなじみBlue Pippinを使ったサイズセレクション
普通は7kbカットオフとか10kbカットオフとかを行なっていますが、ここでは17kbカットオフをした。
17kb未満のサイズのライブラリを捨てて、それ以上の長いライブラリだけを回収するというわけ。
さて、そのようにしてできたライブラリを、4時間と6時間でシークエンスしたら・・・
 リード数は4時間より6時間の方が多いですが、これは偶然でしょう。
リード数は4時間より6時間の方が多いですが、これは偶然でしょう。
平均リード長は、4時間が9kb、6時間が10kb
平均サブリード長は、4時間が8kb、6時間が8.7kb
すごく長いというわけではないけれど、20kb、30kb超えのサブリードも結構ありましたので、これでHGAP3アセンブルを試みた。
 Contig数はどちらも4本
Contig数はどちらも4本
最長Contigはどちらも5.32Mb
6時間Contigの配列でDot Plotを作ってみると、確かに、20kbから30kbの長さのリピートが多く含まれていた。
おおーっ!すごい!
 6時間で作ったContigに、再度サブリードをマップして作られた、カバレッジグラフを見てみると、染色体5.32MbのContigは、60カバレッジ~140カバレッジであることがわかりました。
6時間で作ったContigに、再度サブリードをマップして作られた、カバレッジグラフを見てみると、染色体5.32MbのContigは、60カバレッジ~140カバレッジであることがわかりました。
カバレッジが高い場所は、ORIであるかも知れない。
ほかのContigは、プラズミドかな? これはその道の専門家に調査をお願いしています。
さて、結果としては、4時間でも6時間でも、このゲノムの染色体はつながりました。
ちゃんと精査する余地はまだ残っているとしても。
せっかくなのでもっとすごいリード、サブリードの例もお見せします。
これは別の大腸菌です。
 リード数も6万本、7万本と、さっきの株より多いけど、リード長はもっとすごい。
リード数も6万本、7万本と、さっきの株より多いけど、リード長はもっとすごい。
平均リード長は、4時間が15.6kb、6時間が18.2kb
平均サブリード長も、4時間が13.2kb、6時間が14.4kb
もちろんこのデータでも、染色体ゲノムのアセンブル成功
20kbのリピートも何のその! です。
もちろん、このようにサイズセレクションをシビアにすると、捨てられるDNA量も多いですから、最初に用意すべきDNA量は大変多い(10マイクロ~30マイクログラム)です。
これがネックでしょうね。少量DNAからもこのような長いライブラリを作れたら良いのですが。
本当は10月最初の話題はこれにするつもりだったのですが、新型機械の発表があったもんだから、インパクトに欠けてしまいました。
でも、8月に実際シークエンスをしてみて、アセンブルしてびっくりしたのでせっかくだからシェアします。
(公開にあたりサンプル提供者の許可は得ています)
このサンプル、腸管出血性大腸菌O111は、ゲノムの中に20kb~30kbのリピートが多く、ショートリードではアセンブルがとても難しい。
PacBioでも、一昔前の酵素では、同じように複雑なゲノムであるO157:H7ゲノムは200カバレッジで読んでもContig数は9本でした。
(Koren S., et. al. (2013) Reducing assembly complexity of microbial genomes with single molecule sequencing. Genome Biology, 14:R101 Table 3)
そんなところに、この度、最長Movie時間が4時間から6時間にバージョンアップ!
6時間シークエンスということは、超長いライブラリを作成すれば、長いサブリードが得られ、結果アセンブル結果も改善できるはず。
ということで、
- 40kbライブラリを作製
- サイズセレクションをよりシビアに
- 4時間または6時間でシークエンス
を試しました。
40kbライブラリを作製するには、それなりに長くゲノムを切ることが必要。
今までのG-tubeではなく、Megaruptorという機器を使って切りました。
Megaruptorについてはこちら
この機械で何回かテストカットして、Pippin Pulseに流して確認。
本番カットでゲノムを40kb Shearingしたら、PacBio SMRT bellライブラリ作製へ。
ライブラリができたらおなじみBlue Pippinを使ったサイズセレクション
普通は7kbカットオフとか10kbカットオフとかを行なっていますが、ここでは17kbカットオフをした。
17kb未満のサイズのライブラリを捨てて、それ以上の長いライブラリだけを回収するというわけ。
さて、そのようにしてできたライブラリを、4時間と6時間でシークエンスしたら・・・

平均リード長は、4時間が9kb、6時間が10kb
平均サブリード長は、4時間が8kb、6時間が8.7kb
すごく長いというわけではないけれど、20kb、30kb超えのサブリードも結構ありましたので、これでHGAP3アセンブルを試みた。

最長Contigはどちらも5.32Mb
6時間Contigの配列でDot Plotを作ってみると、確かに、20kbから30kbの長さのリピートが多く含まれていた。
おおーっ!すごい!

カバレッジが高い場所は、ORIであるかも知れない。
ほかのContigは、プラズミドかな? これはその道の専門家に調査をお願いしています。
さて、結果としては、4時間でも6時間でも、このゲノムの染色体はつながりました。
ちゃんと精査する余地はまだ残っているとしても。
せっかくなのでもっとすごいリード、サブリードの例もお見せします。
これは別の大腸菌です。

平均リード長は、4時間が15.6kb、6時間が18.2kb
平均サブリード長も、4時間が13.2kb、6時間が14.4kb
もちろんこのデータでも、染色体ゲノムのアセンブル成功
20kbのリピートも何のその! です。
もちろん、このようにサイズセレクションをシビアにすると、捨てられるDNA量も多いですから、最初に用意すべきDNA量は大変多い(10マイクロ~30マイクログラム)です。
これがネックでしょうね。少量DNAからもこのような長いライブラリを作れたら良いのですが。
2015年10月1日木曜日
PacBio 新型シークエンサー Sequel System
皆さん、驚かれるかも知れませんが、PacBioの新型シークエンサーが発表されました。
 私も全く知らなかった。
私も全く知らなかった。
本当にサプライズ。
PacBio本社でも、知っているひとは限られていたそうです。
ついこの間まで、PacBioの誰もが「PacBioはあの大きな装置、RSIIを、これからも売っていく。小型シークエンサーは出ない」って明言していましたから。
私もそれを信じて、あちこちで「新型装置は出ない」って言ってきたので、結果として嘘を言っていたことになり、申し訳ないです。
とは言っても、MiSeqくらいの大きさ・・・というわけではないようですね。まだでかい。
 横に立っている女性、背が高いです。
横に立っている女性、背が高いです。
装置の大きさは幅x奥行きx高さが、36.5 in x 34 in x 66 in(168 cmくらい)
重さは381kg
今までのRSに比べると確かに小型です。軽乗用車サイズから冷蔵庫サイズに、なった感じ。
デザインは・・・ ちょっと・・・ まあ、いろいろあると思いますがあえて言いません。
プレスリリースはこちら

では今までのRS IIとどう変わったのか?
SMRT Cellが大きくなりました。大きさにして約4倍、ZMWの数は約100万
ここから出力されるリードの本数は、今までと比べておよそ6~7倍
リードの長さやMovie時間はRSIIと変わらない予定です。
1セルあたりの出力塩基数は~5Gb(うまくいけば10Gbも?)
最大16セルまで1ランで使用可能
SMRT Cellが大きくなったということで、今までは8セルが1本のStripにまとまっていましたが、Sequel Systemは4セルで1本です。
これを最大4本、装置にセットすることができます。(合計最大16セル)
Sequel Systemでできることは、
デノボアセンブリ、Iso-Seq、ロングアンプリコン、Methylation解析、というふうにRSIIと同じです。
サンプル調製も、一部はRSIIと同じキットを使い、一部は特別のものを使う、というふうに分かれています。
Sequel Systemを使えば、うまくいけば、30xのヒトゲノム・90Gbを、1回のランで得ることも可能。
それも平均10kbの超ロングリードで!!


今年のASHG(アメリカ人類遺伝学会)の大きな目玉になることは間違いない!
そもそも、どうしてこんな製品が突然出てきたのでしょうか?
ご存知の方もいると思いますが、PacBioはRocheと共同で、Roche向けの診断用NGS装置を開発するという契約がありました。以前ここにも書きました。
この装置も、Roche次世代シークエンサーの開発の過程で作られたものだそうです。
極秘裏に開発されたのも仕方が無い。
こちらSequel Systemは、Research Onlyなので、Roche向け診断用NGSとは、別物です。
診断向けNGSについては、Rocheさんにお問い合わせ下さいね。
肝心のデータですが、実は、まだオープンにできるものはありません。
現在、本社でサンプル調製の最適化と共に、良いデータ出しをしているところだと思います。
乞うご期待!
価格は来週決まりますので、お問い合わせはトミーデジタルまでお気軽に!
しかし、当然ですが、気になるのは今後RSIIの運命はどうなるのか? ということですよね。
先ほどのプレスリリースによると、RSIIの試薬の開発・サポートは続くとのことです。
これは信じる。
それに、RSIIは販売してから歴史があるので、トラブルシュートの経験がある。
新しいSequel Systemよりは安定してデータを出してくれるでしょう。
しばらくは共存していくと思います。
来週アメリカに行くので、いろいろ突っ込んで聞いてきます。
あーあ、それにしても、あちこちで「新型機械は出ない!」って言ってきたなあ。 やばい。
サプライズとしては良いかも知れないけれど。
出るとわかっていたらもっと嬉しかったかも知れない。そう思う社員は、PacBio本社の中にも多いと思う。
まあ、この業界では良くある話ですけどねえ。
良いニュースとして、前向きに考えることにしました!

本当にサプライズ。
PacBio本社でも、知っているひとは限られていたそうです。
ついこの間まで、PacBioの誰もが「PacBioはあの大きな装置、RSIIを、これからも売っていく。小型シークエンサーは出ない」って明言していましたから。
私もそれを信じて、あちこちで「新型装置は出ない」って言ってきたので、結果として嘘を言っていたことになり、申し訳ないです。
とは言っても、MiSeqくらいの大きさ・・・というわけではないようですね。まだでかい。

装置の大きさは幅x奥行きx高さが、36.5 in x 34 in x 66 in(168 cmくらい)
重さは381kg
今までのRSに比べると確かに小型です。軽乗用車サイズから冷蔵庫サイズに、なった感じ。
デザインは・・・ ちょっと・・・ まあ、いろいろあると思いますがあえて言いません。
プレスリリースはこちら

では今までのRS IIとどう変わったのか?
SMRT Cellが大きくなりました。大きさにして約4倍、ZMWの数は約100万
ここから出力されるリードの本数は、今までと比べておよそ6~7倍
リードの長さやMovie時間はRSIIと変わらない予定です。
1セルあたりの出力塩基数は~5Gb(うまくいけば10Gbも?)
最大16セルまで1ランで使用可能
SMRT Cellが大きくなったということで、今までは8セルが1本のStripにまとまっていましたが、Sequel Systemは4セルで1本です。
これを最大4本、装置にセットすることができます。(合計最大16セル)
Sequel Systemでできることは、
デノボアセンブリ、Iso-Seq、ロングアンプリコン、Methylation解析、というふうにRSIIと同じです。
サンプル調製も、一部はRSIIと同じキットを使い、一部は特別のものを使う、というふうに分かれています。
窒素ガスはRSII同様、必要です。
この機械の、前面の黒いところは、スライドして下に降りるようになっています。
すると、中からロボットと試薬セットを置く台が現れます。
ここでユーザが、SMRT Cellをセットしたり、キットやチップを置いたりします。
スライドを閉めてから、上段右のタッチパネルで操作します。
Real Timeでシークエンスが始まり、データはこの装置の下のほうにあるサーバで、ベースコールされます。
ベースコールされたデータは、ユーザのサーバに転送されるか、USB3.0でユーザが抽出します。
データ解析はRSIIと同様、SMRT Analysis 3.0 を使います。
あ、SMRT Analysisは次に3.0になります。ちなみに3.0はRSIIのデータも解析できますのでご安心を。
それも平均10kbの超ロングリードで!!


今年のASHG(アメリカ人類遺伝学会)の大きな目玉になることは間違いない!
そもそも、どうしてこんな製品が突然出てきたのでしょうか?
ご存知の方もいると思いますが、PacBioはRocheと共同で、Roche向けの診断用NGS装置を開発するという契約がありました。以前ここにも書きました。
この装置も、Roche次世代シークエンサーの開発の過程で作られたものだそうです。
極秘裏に開発されたのも仕方が無い。
こちらSequel Systemは、Research Onlyなので、Roche向け診断用NGSとは、別物です。
診断向けNGSについては、Rocheさんにお問い合わせ下さいね。
肝心のデータですが、実は、まだオープンにできるものはありません。
現在、本社でサンプル調製の最適化と共に、良いデータ出しをしているところだと思います。
乞うご期待!
価格は来週決まりますので、お問い合わせはトミーデジタルまでお気軽に!
しかし、当然ですが、気になるのは今後RSIIの運命はどうなるのか? ということですよね。
先ほどのプレスリリースによると、RSIIの試薬の開発・サポートは続くとのことです。
これは信じる。
それに、RSIIは販売してから歴史があるので、トラブルシュートの経験がある。
新しいSequel Systemよりは安定してデータを出してくれるでしょう。
しばらくは共存していくと思います。
来週アメリカに行くので、いろいろ突っ込んで聞いてきます。
あーあ、それにしても、あちこちで「新型機械は出ない!」って言ってきたなあ。 やばい。
サプライズとしては良いかも知れないけれど。
出るとわかっていたらもっと嬉しかったかも知れない。そう思う社員は、PacBio本社の中にも多いと思う。
まあ、この業界では良くある話ですけどねえ。
良いニュースとして、前向きに考えることにしました!
2015年9月23日水曜日
Flatwormは再生医療に役立つか?
この仕事をしていると、いろんな生物を研究しているひとと出会うんですね。
初めて知る生物の名前や、今まで誤解していた生物など、結構あるものです。
例えば、ナメクジウオ
名前から、魚なのかと思っていましたが、とんでもない。
頭索動物、という仲間で、脊索はあるけれど脊椎は無い。
つまり脊椎動物と無脊椎動物の分岐点に位置する動物というわけで、進化の研究に重要な生物だそうです。
ほかにもたくさんありますが、それはまた今度の機会に。
今日は、Flatworm(扁形動物)という生物の話です。
プラナリア、ヒラムシ、サナダムシの仲間というだけで、気持ち悪ぅー、と思ったあなた。
あなたはこの生物がどんなにすごいかを知れば、絶対考えを変えるでしょう。
切っても体が再生するんですよ!
ザリガニは足が再生しますがそんなものではない!
 |
| http://www.macgenome.org/animal.html より |

Wasik et al., (2015) Genome and transcriptome of the regenerationcompetent flatworm, Macrostomum lignano. PNAS
M. linganoという生物、こいつは切っても完全に再生するそうです。
細胞の自己再生、組織の再生の研究にはもってこいというわけ。
そこで、Cold Spring Harbor Laboratory のMicheal Schatz博士らのグループは、この生物のゲノムとトランスクリプトームを読んで、組織再生の謎に迫りました。
PacBioが活躍したのは、ゲノムアセンブリ。
M. linganoのゲノムはK-mer (23-mer) 解析から700Mbと想定。
全ゲノムの70%がリピートでトランスポゾン配列が多く、2n=8本の染色体はChromosomal Duplicationもあるとされ、当然ながらショートリードではまったくつながらない。
そこで、PacBioロングリードの登場!
ゲノム配列を10ug用意して10kbをターゲットにg-Tubeで切断、その後SMRT bellライブラリを作製し、6kb~15kbでサイズセレクションしたあと、P4C2またはP5C3でシークエンス。
試薬のバージョンからして、実験は昨年されたのでしょうね。
130x分のシークエンスデータを得た後、エラー補正を施し、実際アセンブルに用いたのは10kb以上の補正後リード21x分。
そうして、N50=64kbのContigを得た。64kbというのはこれまでの222bp(ショートリードでのアセンブリ)に比べると、大きな進歩。
次に彼らが行なったのは遺伝子アノテーションと、発現解析。
発現解析はIlluminaで行なっていますが、まあ、これはこれとして、リファレンスContigができたおかげですから。
山中ファクターとしても有名な、Oct4/Pou5f1、Nanog、Klf4、c-Myc、Sox2遺伝子は、組織再生の重要ファクターです。
この生物では、Sox2以外の遺伝子発現は見られなかったものの、哺乳類の幹細胞メインテナンスに必要な、Jak-Stat、Wnt、MAPKパスウェイは保存されていたそうです。
そのほかにも、組織が再生される過程を示すパスウェイや特定の遺伝子発現などが、哺乳類のそれと、似ているところ、似ていないところ、が示されました。
まだわからないことは多いけれど、なるほど、気持ち悪い生物も結構我々と共通するところあるんだなあ、と思いましたね。
気持ち悪い生物、と言ったら失礼か。
個人的には、できればIso-SeqをP6C4でもやって欲しい。
もうやっていたりして。
2015年9月19日土曜日
PacBioでHLAシークエンス
先日、水戸で「日本組織適合性学会大会」があり、行ってきました。
以前からPacBioでHLA遺伝子をシークエンスしている先生の発表があったのと、この業界についてはほとんど素人だったので、勉強もかねて。学会についてはこちら
この学会では、輸血・臓器・造血といった、移植全般の「組織適合性」に関して、大学研究者から医療関係者、日本赤十字、現場のティシュータイパー、といった方々が集まって最新の技術を発表したり、現場の問題点を提起したり、活発に議論が交わされていました。
企業からもタイピング受託会社や、タイピング装置やキットの輸入商社、メーカーなどが参加。
学会の規模は、そうですね、実感としては「ゲノム微生物学会」くらいの大きさかな。
さて、HLAについて、もし基礎的なバックグラウンドを知りたいかたは、先ほどの学会ホームページのここがとても詳しいです。
私もこのページで勉強しました。
ざっくり言うと、移植のときなどにドナーとレシピアントとの間で、適合するかどうか、は、HLAという遺伝子のSNPの型で決めるんです。
ひとつの遺伝子にSNPがいくつもあって、その組み合わせで、何千通りというハプロタイプができる。
HLAはA、B、C、DRB、DQB、DPBなどなどいくつかあり、タイピングに用いられる遺伝子はだいたい決まっています。
その遺伝子のSNP型を、ビーズ、PCR、またはシークエンスで判定するということ。
今、HLAタイピングの主流はビーズです。
その理由は、①簡単、②低コスト、③世界中でバリデートされている
から。
ビーズで検出すると、同じ遺伝子上でも離れた場所にある2つのSNPのハプロタイプはわからない。
これをAmbiguity問題、といいます。
シークエンス法に比べると、精度は落ちる。
シークエンス法の場合、先ず一旦遺伝子の全長、または変異の高い場所をPCR増幅します。
プライマーはもちろん保存された領域に設定される。
このプライマーでちゃんと増えたら、次に断片化する。
断片化した後、シークエンスする場所を濃縮するか、あるいはそのままシークエンスする。
あれ? 何で断片化するの?
と思ったかた、あなたの頭はPacBio向きです。
PCRで増幅したHLA遺伝子は、だいたい4kbから10kbかそれより少し長いくらい。
PacBioならそのまま読めますよね。
そうです。読めます。
断片化するのは、ショートリードのときだけ。
NGSとHLAタイピングをもっと詳しく知りたい!という方はこのレビューがおすすめ

Hosomichi et al., (2015) The impact of next-generation sequencing technologies on HLA research. J of Human Genetics.
シークエンスされたデータは、IMGT/HLAデータベースに登録されている各HLA遺伝子の配列にマッピングされます。マッピングのアルゴリズムは、ツールごとにいろいろあるようです。
以前からPacBioでHLA遺伝子をシークエンスしている先生の発表があったのと、この業界についてはほとんど素人だったので、勉強もかねて。学会についてはこちら
この学会では、輸血・臓器・造血といった、移植全般の「組織適合性」に関して、大学研究者から医療関係者、日本赤十字、現場のティシュータイパー、といった方々が集まって最新の技術を発表したり、現場の問題点を提起したり、活発に議論が交わされていました。
企業からもタイピング受託会社や、タイピング装置やキットの輸入商社、メーカーなどが参加。
学会の規模は、そうですね、実感としては「ゲノム微生物学会」くらいの大きさかな。
さて、HLAについて、もし基礎的なバックグラウンドを知りたいかたは、先ほどの学会ホームページのここがとても詳しいです。
私もこのページで勉強しました。
ざっくり言うと、移植のときなどにドナーとレシピアントとの間で、適合するかどうか、は、HLAという遺伝子のSNPの型で決めるんです。
ひとつの遺伝子にSNPがいくつもあって、その組み合わせで、何千通りというハプロタイプができる。
HLAはA、B、C、DRB、DQB、DPBなどなどいくつかあり、タイピングに用いられる遺伝子はだいたい決まっています。
その遺伝子のSNP型を、ビーズ、PCR、またはシークエンスで判定するということ。
今、HLAタイピングの主流はビーズです。
その理由は、①簡単、②低コスト、③世界中でバリデートされている
から。
ビーズで検出すると、同じ遺伝子上でも離れた場所にある2つのSNPのハプロタイプはわからない。
これをAmbiguity問題、といいます。
シークエンス法に比べると、精度は落ちる。
 |
| GenDX社のウェブサイトから |
プライマーはもちろん保存された領域に設定される。
このプライマーでちゃんと増えたら、次に断片化する。
断片化した後、シークエンスする場所を濃縮するか、あるいはそのままシークエンスする。
あれ? 何で断片化するの?
と思ったかた、あなたの頭はPacBio向きです。
PCRで増幅したHLA遺伝子は、だいたい4kbから10kbかそれより少し長いくらい。
PacBioならそのまま読めますよね。
そうです。読めます。
断片化するのは、ショートリードのときだけ。
NGSとHLAタイピングをもっと詳しく知りたい!という方はこのレビューがおすすめ

Hosomichi et al., (2015) The impact of next-generation sequencing technologies on HLA research. J of Human Genetics.
HLAタイピングの方法、ソフトウェアの種類、長所短所などがまとめられています。
有償ソフトウェアなどは各社独自のマッピング方法を開発して、精度を上げています。
そして、アレルごとにコンセンサスを出して、アレルの型を決める
ここはPacBioでのHLAシークエンスに一番ノウハウがあると思いますが、いかんせん、企業なので情報はあまり出てこない。
では、イギリスのAnthony Nolanはどうかな?ここは公共機関。論文もあります。
 Mayor et al., (2015) HLA Typing for the Next Generation. PLOS One.
Mayor et al., (2015) HLA Typing for the Next Generation. PLOS One.

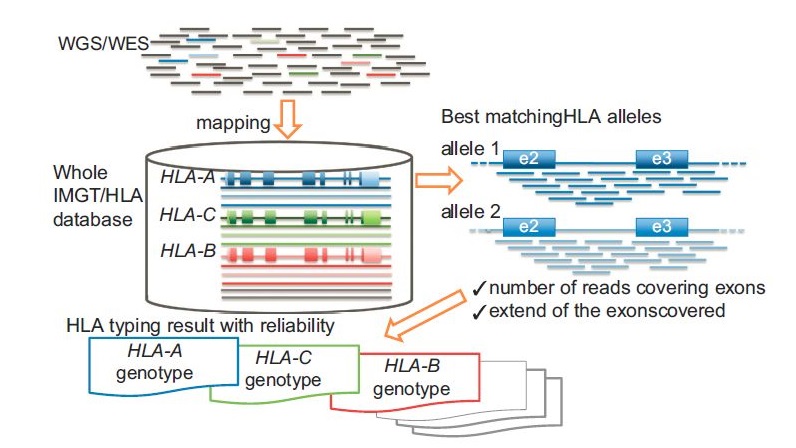 |
| Hosomichi et al. (2015) |
PacBioの場合も、長いリードで遺伝子全体を読みますが、そのあとデノボでクラスタリングをつくり、アレルごとにコンセンサス配列を作ります。
先にデータベースにマッピングするのではなく、アレルごとの遺伝子全体のコンセンサス配列をつくってから、データベースを参照して型を判定するのです。
(正確には、遺伝子全体を読むのはClass Iと呼ばれる比較的短い、とはいっても5kbくらい、の遺伝子。Class IIと呼ばれる遺伝子群は、10kb以上あるが、それくらい長いとPCRすること自体が難しい。そこで遺伝子の一部を増幅することが多い)
このように、NGS、特にPacBioで読めば、Ambiguity無しに、正確なHLA型を判定することが可能です。
でも、タイピングにかかるコストは若干高め。
ライブラリ作製にかかる手間は、ビーズ法に比べれば、かかる。これは認めます。
では、どういう場合に、PacBioがHLAタイピングに使用されているのか?
アメリカには、Histogenetics社という、HLAタイピングの専門会社があります。
この会社はMiSeqを47台、PacBioを1台保有し(もちろんキャピラリーはたっくさん保有)、Sequence Based Typing (SBT)をガンガンやっています。
顧客は主にNMDP(National Marrow Donor Program)のようなバンク。
MiSeqを使ったタイピングでもキャピラリーを使ったタイピングでも、データベースに合わなかったり新規の可能性があるような場合、PacBioの登場。
昨年はまだ慣れていなかったテクニシャンも、もうPacBioかんたーん!って言っているそうです(Histogenetics社のCEO曰く)。
タイピングの方法も、自分たちで作ったデータベースに独自のプログラムを使って、行なっているとのこと。ここはPacBioでのHLAシークエンスに一番ノウハウがあると思いますが、いかんせん、企業なので情報はあまり出てこない。
では、イギリスのAnthony Nolanはどうかな?ここは公共機関。論文もあります。

新しいアレルも発見されています

Hayward et al., (2015) The novel HLA-B*44 allele, HLA-B*44:220, identified by Single Molecule Real-Time DNA sequencing in a British Caucasoid male. Tissue Antigens.
人類遺伝学会などでも関連する発表がありそうな予感。
PacBioの可能性が試されるときです!
2015年8月17日月曜日
ところで最近cDNAシークエンスはどうなった
アイソフォームシークエンス、通称 Iso-Seq
これは以前もこのブログで紹介しました
ところで、良くある質問が、どれくらい読んだら十分か、というもの。
転写量が低いアイソフォームも高いものも、まんべんなく検出するには、何セル読んだら良いのか?
簡単なようで難しい問題です。
これは逆に出力から考えた方が良さそうです。
1つのSMRT Cellから出力されるリード数は、およそ6万本。
1本1本が独立のアイソフォーム配列由来です。
ReadsOf Insert、別名CCSですが、これがちゃんと全長cDNAをカバーしているかどうかが大事です。
ここで全長というのは、逆転写酵素で転写産物を復元した後、PCR増幅するときのPCRプライマー配列が、シークエンスされた後のReadsOf Insertで、5’側と3’側にちゃんとあることを言います。
つまり、長いアイソフォームほど、全長読まれる確率は低くなる
設定するReads Of Insetのパス数は、アイソフォーム配列の精度に影響する
では、出力されるReadsOfInsertのうち、どれくらいが全長読まれたものなのか?
先月のユーザーミーティングでは、いつくかIso-Seqの発表もありました。
その中のひとつでは、3パターンでサイズセレクションをしていて、そのうち完全長cDNAだった割合は、
というふうに、転写産物の長さが長くなるほど、完全長アイソフォームの数は少なくなりました。
当然といえば当然。
他の発表でも、サイズセレクションのデータは無いけれど、8セル使ったIso-Seqの実験で完全長は21万本。
64セル読んだときは完全長cDNAは100万本(全体のリード数は470万本)だったそうです。
このような数字が、自分の目的に合うかどうか?
で、計算してはいかがでしょうか。
しかし、完全長cDNAといっても、もしかすると5’側の配列が欠けていることがあるかもしれません。
これは逆転写酵素Takara-Clontech SMARTerが、最初に転写産物の5’キャッピングをしないことが原因です。
5’側まで行かなくてもcDNAが完成してしまうため、ある程度、完全長では無いcDNAができてしまいます。
その後PCR増幅するときに使う5’と3’のプライマー配列が、シークエンスで読めていれば、Iso-Seqでは完全長cDNAと言います。
ここ、気をつけて下さい。
もうひとつ、その昔、Iso-Seqが開発途中だった3年前の話です。
転写ノーマライゼーションというものがありました。
これは、転写量が高いアイソフォームばかり読めてしまって、転写が低い産物がなかなか読めないことを防ぐために考えられたプロトコルです。
カムチャッカカニから抽出したDuplex-specific nuclease(DSN)を使用した方法で、原理としては以下のようなもの。
これは以前もこのブログで紹介しました
ところで、良くある質問が、どれくらい読んだら十分か、というもの。
転写量が低いアイソフォームも高いものも、まんべんなく検出するには、何セル読んだら良いのか?
簡単なようで難しい問題です。
これは逆に出力から考えた方が良さそうです。
1つのSMRT Cellから出力されるリード数は、およそ6万本。
1本1本が独立のアイソフォーム配列由来です。
ReadsOf Insert、別名CCSですが、これがちゃんと全長cDNAをカバーしているかどうかが大事です。
ここで全長というのは、逆転写酵素で転写産物を復元した後、PCR増幅するときのPCRプライマー配列が、シークエンスされた後のReadsOf Insertで、5’側と3’側にちゃんとあることを言います。
つまり、長いアイソフォームほど、全長読まれる確率は低くなる
設定するReads Of Insetのパス数は、アイソフォーム配列の精度に影響する
では、出力されるReadsOfInsertのうち、どれくらいが全長読まれたものなのか?
先月のユーザーミーティングでは、いつくかIso-Seqの発表もありました。
その中のひとつでは、3パターンでサイズセレクションをしていて、そのうち完全長cDNAだった割合は、
- 1-2kb:50%(1セル6万本出力と仮定すると、3万本)
- 2-3kb:30%(同18,000本)
- 3kb-:20%(同12,000本)
というふうに、転写産物の長さが長くなるほど、完全長アイソフォームの数は少なくなりました。
当然といえば当然。
他の発表でも、サイズセレクションのデータは無いけれど、8セル使ったIso-Seqの実験で完全長は21万本。
64セル読んだときは完全長cDNAは100万本(全体のリード数は470万本)だったそうです。
このような数字が、自分の目的に合うかどうか?
で、計算してはいかがでしょうか。
しかし、完全長cDNAといっても、もしかすると5’側の配列が欠けていることがあるかもしれません。
これは逆転写酵素Takara-Clontech SMARTerが、最初に転写産物の5’キャッピングをしないことが原因です。
5’側まで行かなくてもcDNAが完成してしまうため、ある程度、完全長では無いcDNAができてしまいます。
その後PCR増幅するときに使う5’と3’のプライマー配列が、シークエンスで読めていれば、Iso-Seqでは完全長cDNAと言います。
ここ、気をつけて下さい。
もうひとつ、その昔、Iso-Seqが開発途中だった3年前の話です。
転写ノーマライゼーションというものがありました。
これは、転写量が高いアイソフォームばかり読めてしまって、転写が低い産物がなかなか読めないことを防ぐために考えられたプロトコルです。
カムチャッカカニから抽出したDuplex-specific nuclease(DSN)を使用した方法で、原理としては以下のようなもの。
- 一度cDNAをDenatureしたあと、Renatureする→ Abundantな転写産物ほど二本鎖に戻りやすいはず
- 二本鎖DNAを特異的にHydrolyzeする酵素(DSN)で処理する→ Abundantな転写産物ほど優先的に分解される
- 転写レベルが低かったcDNAが分解されずに残る
→ これでライブラリを作ることで、レアなcDNAも少ないセル数でシークエンスすることができるし、高発現だったcDNAは、2のところで分解されてライブラリにならないはず
しかーし、結局公式プロトコールにはならなかった。理由は以下の通り
- cDNAを一旦Denatureした後、二本鎖にする段階で、長いcDNAほど、同じ配列(ドメイン配列など)を有する他のアイソフォームと非特異的に二本鎖を形成する確率が高くなる
- 長いcDNAは、それが例えレアな転写産物であっても、非特異的Renatureを作りやすいことで、結果DSN分解(hydrolysis)されてしまう
- ノーマライゼーションステップには、追加でPCR増幅が必要なため、さらに増幅バイアスが生じる(Iso-Seqは2回、PCR増幅しますので、ノーマライズするときは合計3回のPCRが必要になる)
これらを考えて、海外のあるユーザは、2kb未満の比較的短いcDNAに対してのみ、ノーマライズ処理をしているそうです。
短ければ非特異的Renatureはある程度防げるだろう、という考えです(増幅バイアスはかかりますが)。
とは言うものの、PacBioとして公式に勧めているプロトコルではありません。自己責任です。
酵素自体はEvrogen社で販売されているようですね。
じゃあ今はどうなんだ? と聞かれそうですが、今もノーマライゼーションは公式プロトコルにはありません。
転写産物の高いものだけを、読む前に減らす方法は難しいのかな。
登録:
投稿 (Atom)