年末といえばジャンボ宝くじ! 宝くじといえば当選確率、ということで、無理やりですが今日は確率の話です。
PacBioのリードには、エラーがランダムに入ります。
その割合は前にも書きましたが13~15%くらい。
ということでリード1本あたりの精度がおよそ85%、というふうになるわけです。
で、この精度を上げるために、カバレージを増やします。

でも精度85%のリードを重ねただけで、そんなに正確性が上がるのはなぜ?
という疑問を持たれる方もいるかもしれませんね。
高校の確率・統計で苦しんだ私がご説明致します。
話を単純にするために、正しい塩基を多数決で決めることとします。
また、PacBioのエラー率は実際より少し高めの、16.7%とします。
つまり、16.7% = 1/6 の確率でランダムに間違える。
リードが1本の場合、間違える確率は1/6 です。 ここまではOK?
次にリードが2本の場合、
1本が間違える確率 P(1 of 2) + 2本とも間違える確率 P(2 of 2) が、間違える確率です。
ちょっと格好良く言うと、1/6の確率で起こる事象があり、それを2回試したとき、1回以上起こる確率です。
サイコロを2回振って、1が1回以上出る確率と同じですね。
リードが3本の場合、
2本が間違える確率 P(2 of 3) +3本とも間違える確率 P(3 of 3) が、間違える確率です。
同じく格好良く言うと、1/6の確率で起こる事象があり、それを3回試したとき、2回以上起こる確率です。
サイコロを3回振って、1が2回以上出る確率と同じです。

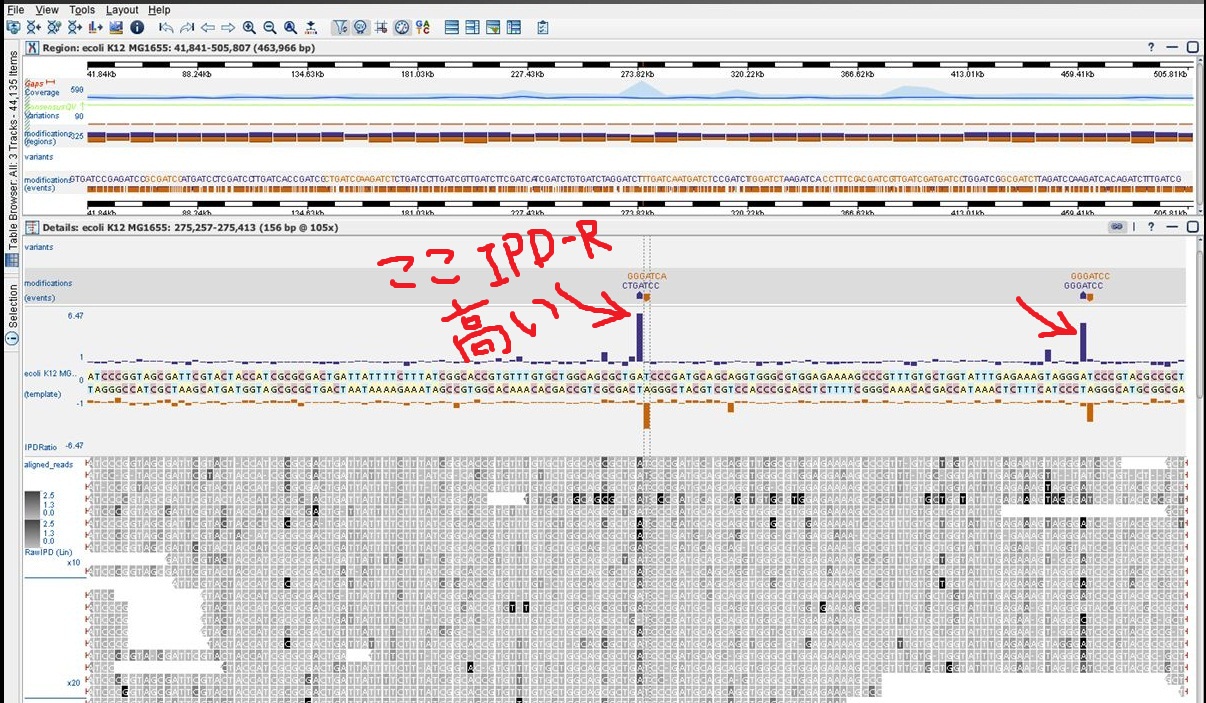
絵にするとこんな感じで、上から1本のとき、2本のとき、3本のとき、の「間違い」リードの組み合わせです。間違いリードは×印です。
さて、ではリード4本のとき、
2本が間違える確率 P(2 of 4) + 3本が間違える確率 P(3 of 4) + 4本とも間違える確率 P(4 of 4)
=>1/6の確率で起こる事象があり、それを4回試したとき、2回以上起こる確率
(サイコロを4回振って1が2回以上出る確率)
リード5本のときは?
3本が間違える確率 P(3 of 5) + 4本が間違える確率 P(4 of 5) + 5本とも間違える確率 P(5 of 5)
=>1/6の確率で起こる事象があり、それを5回試したとき、3回以上起こる確率
(サイコロを5回振って1が3回以上出る確率)
=>1/6の確率で起こる事象があり、それを5回試したとき、3回以上起こる確率
(サイコロを5回振って1が3回以上出る確率)

1回の試行で特定の事象が起きる確率がpのとき、n回の試行で特定の事象がm回起きる確率Pは、

です。 これ、関数電卓なら頑張れば計算できますけど、今は便利なサイトがあるもので、
こちら、その名もke!san http://keisan.casio.jp/#menu
ホーム/ 計算応用集/ 確率・統計/ サイコロの目の出る確率 で、上の式の計算が簡単にできます。そうすると、先ほどのリードの場合は以下のような数字になります。


ここまで 0.167 -> 0.306 -> 0.074 -> 0.132 -> 0.035 というふうに間違い確率「エラー率」は上がり下がりしながら全体としては下がってきました。
これを、n - > 50 までやってみるとどうなるか?

11カバレージでQV23、19カバレージでQV34、30カバレージでQV46、40カバレージでQV59
このグラフ、上がったり下がったりしながらも、0に近似されていく、・・・ 美的ですね。
これを、1000bpのシュミレーションデータで検証した方がおります。

Sergey Koren et al., “Hybrid error correction and de novo assembly of single-molecule sequencing reads” Nature Biotechnology (2012)
先のグラフと非常に似ていますね。
ランダムであれば、リードを重ねれば全体としてのエラー率は確実に下がっていくのです。
どうでしょうか?
85%精度のリードでも、重ねると100%に近くなる、というのは本当なんですよ。
と、今日はデータの精度を、確率論で説明を試みました。
数学者からはツッコミが入りそうですけれど。
どうでしょうね。